
La sindrome di DiGeorge, causata da una microdelezione nel cromosoma 22 (regione 22q11.2), è la più comune sindrome da microdelezione nell’essere umano e colpisce circa 1 neonato su 4.000. Si manifesta con un quadro clinico molto variabile che può includere cardiopatie congenite, deficit immunitari, ipocalcemia, caratteristiche facciali tipiche e difficoltà del neurosviluppo. La diagnosi precoce è fondamentale per avviare tempestivamente la gestione multidisciplinare e migliorare la qualità di vita dei pazienti.
La storia della sindrome di DiGeorge
La sindrome prende il nome da Angelo DiGeorge, un pediatra ed endocrinologo statunitense di origini italiane che nel 1965 pubblicò la prima descrizione sistematica della condizione. DiGeorge osservò in un gruppo di neonati un quadro ricorrente e riconoscibile: malformazioni cardiache, convulsioni nelle prime ore di vita da ipocalcemia, infezioni gravi e ripetute per assenza del timo, e tratti facciali caratteristici comuni a tutti i bambini colpiti. L’intuizione clinica fu importante, ma la causa biologica rimase oscura per quasi trent’anni: all’epoca non esistevano strumenti molecolari in grado di identificare alterazioni submicroscopiche del DNA.
La svolta arrivò nel 1992, quando l’introduzione della tecnica FISH (Fluorescence In Situ Hybridization) permise di identificare, nella maggioranza dei pazienti con il quadro descritto da DiGeorge, una microdelezione nel braccio lungo del cromosoma 22. Fu la prima conferma molecolare che dietro quella costellazione di sintomi apparentemente eterogenei si celava un difetto genetico preciso e localizzabile. Nel frattempo, altri clinici avevano descritto in modo indipendente condizioni che, con il senno di poi, erano la stessa malattia vista da angolature diverse. Nel 1976 il cardiologo giapponese Takao aveva segnalato una serie di pazienti con cardiopatie conotruncali e tratti facciali peculiari, coniando il termine “cono-truncal anomaly face syndrome”.
Due anni dopo, nel 1978, il chirurgo plastico americano Robert Shprintzen aveva descritto una “nuova sindrome” caratterizzata da anomalie del palato, cardiopatia congenita e una facies riconoscibile, che aveva chiamato sindrome velo-cardio-facciale. Né i pazienti di Takao né quelli di Shprintzen presentavano le alterazioni del timo e delle paratiroidi che DiGeorge aveva posto al centro della sua descrizione originale, il che aveva alimentato per anni il dibattito se si trattasse davvero della stessa malattia.
Come descritto dalla dottoressa Caterina Cancrini nel Quaderno scientifico AIDel22 sulla sindrome da delezione 22q11.2, il dubbio fu risolto con le analisi molecolari: in oltre l’80% dei casi descritti da tutti e tre i clinici era presente la stessa delezione sul cromosoma 22. La comunità scientifica decise quindi di unificare tutte le denominazioni sotto un unico termine diagnostico, sindrome da delezione 22q11.2, che rispecchia il difetto molecolare comune e supera le distinzioni storiche legate alla variabilità clinica. Nomi come DiGeorge, Shprintzen e CATCH22 restano in uso nella letteratura e nel linguaggio clinico, ma oggi descrivono tutti la stessa condizione di fondo.
Cos’è la sindrome di DiGeorge
La sindrome di DiGeorge è una condizione genetica congenita causata dalla perdita di un piccolo segmento di materiale genetico sul braccio lungo del cromosoma 22, nella regione identificata come 22q11.2. Nel corso dei decenni, la stessa condizione è stata descritta con nomi diversi dai clinici che ne osservavano le diverse manifestazioni: sindrome velo-cardio-facciale (o sindrome di Shprintzen), sindrome della facies tronco-conale, sindrome CATCH22 (un acronimo che richiama i difetti cardiaci, le anomalie facciali, l’aplasia timica, la palatoschisi e l’ipocalcemia). Oggi tutte queste denominazioni sono riunite sotto un unico termine diagnostico, sindrome da delezione 22q11.2 (22q11.2DS), che riflette il difetto molecolare comune a tutte le presentazioni cliniche.
La ragione per cui questa condizione si presentava con facce così diverse non è casuale; infatti, il fenotipo, cioè l’insieme delle caratteristiche fisiche e cliniche che la malattia produce, è estremamente variabile, anche all’interno della stessa famiglia. Persone con la stessa delezione possono avere sintomi gravissimi o quasi impercettibili. Questa variabilità ha reso storicamente difficile il riconoscimento clinico e continua a rappresentare una delle principali sfide diagnostiche.
Secondo i dati di Orphanet (Il portale delle malattie rare e dei farmaci orfani), la prevalenza alla nascita è stimata tra 1 su 4.500 e 1 su 10.000, con alcune stime più recenti, basate su studi di screening neonatale con metodiche TREC, che collocano la prevalenza intorno a 1 neonato su 2.148 (StatPearls, NCBI, 2025). La sindrome colpisce maschi e femmine in egual misura e non mostra predisposizioni geografiche specifiche.
La causa genetica: cosa succede sul cromosoma 22
Nel 90% circa dei casi, la delezione origina de novo, cioè compare per la prima volta nell’individuo affetto senza essere stata ereditata da nessuno dei genitori. La causa del fenomeno è una ricombinazione meiotica non allelica durante la formazione dei gameti (spermatozoi o ovociti). Sequenze di DNA molto simili tra loro, chiamate low copy repeats (LCR22) e presenti in più copie lungo la regione 22q11.2, si appaiano in modo scorretto durante la meiosi e producono come conseguenza la perdita del segmento compreso tra due di queste ripetizioni. Il risultato è una delezione che, nella maggioranza dei casi, misura circa 3 milioni di coppie di basi (3 Mb).
Nel restante 6-10% dei casi, la delezione è ereditata da un genitore portatore, spesso non diagnosticato perché paucisintomatico. La trasmissione segue un meccanismo autosomico dominante: quando uno dei genitori è portatore della delezione, ogni figlio ha il 50% di probabilità di ereditarla. Questo dato ha implicazioni dirette sulla consulenza genetica familiare.
Tra i geni contenuti nella regione deleta, il più studiato è TBX1 (T-box transcription factor 1), considerato il principale contributore genetico del fenotipo classico. TBX1 è un fattore di trascrizione espresso durante l’embriogenesi nell’apparato faringeo e nel mesenchima circostante, dove regola lo sviluppo delle strutture derivate dal terzo e quarto arco e tasca faringea. La sua aploinsufficiena, cioè la riduzione a una sola copia funzionale del gene, contribuisce in modo determinante ai difetti cardiaci conotruncali, dell’ipoplasia timica e paratiroidea e delle anomalie craniofacciali tipiche della sindrome.
Un secondo gene rilevante, DGCR8, è coinvolto nella regolazione dei microRNA e contribuisce al profilo neuropsichiatrico della condizione; un terzo, COMT, codifica per la catecol-O-metiltransferasi, che metabolizza la dopamina e altre catecolamine, è stato implicato nell’aumento del rischio di disturbi psichiatrici e disfunzioni cognitive osservato nei soggetti affetti come spiegato da Sushma et al. 2025.
Come si sviluppa la Sindrome di DiGeorge durante l’embriogenesi
Capire quando e come la delezione produce i suoi effetti richiede di tornare alle prime settimane di sviluppo embrionale. Durante l’embriogenesi precoce, le cellule della cresta neurale cardiaca migrano dalla regione dorsale del tubo neurale verso l’apparato faringeo e partecipano alla formazione del tratto di efflusso cardiaco, dell’arco aortico, del timo, delle ghiandole paratiroidi e delle strutture craniofacciali. Il gene TBX1, espresso nei tessuti dell’apparato faringeo, è essenziale per coordinare i segnali che regolano questa migrazione e differenziazione. Quando TBX1 è disponibile in quantità dimezzata, il processo si altera: le arterie dell’arco faringeo non si formano correttamente, i cuscinetti dell’efflusso cardiaco non si fondono nel modo previsto, le paratiroidi e il timo non si sviluppano adeguatamente.
Il fatto che TBX1 influenzi l’espressione di un ampio numero di geni – fino a quasi 2.000 secondo studi di trascrittomica (Frontiers in Genetics, 2019) – durante questa fase critica dello sviluppo contribuisce a spiegare perché la sua aploinsufficiena produca effetti così ampi e multisistemici. Spiega anche, almeno in parte, perché lo stesso difetto genetico dia manifestazioni cliniche così diverse: la variabilità del fenotipo dipende da modificatori genetici presenti sugli altri cromosomi, da varianti nel secondo allele 22q11.2 non deleto e da fattori ancora non completamente compresi.
Il quadro clinico della Sindrome di DiGeorge: manifestazioni principali
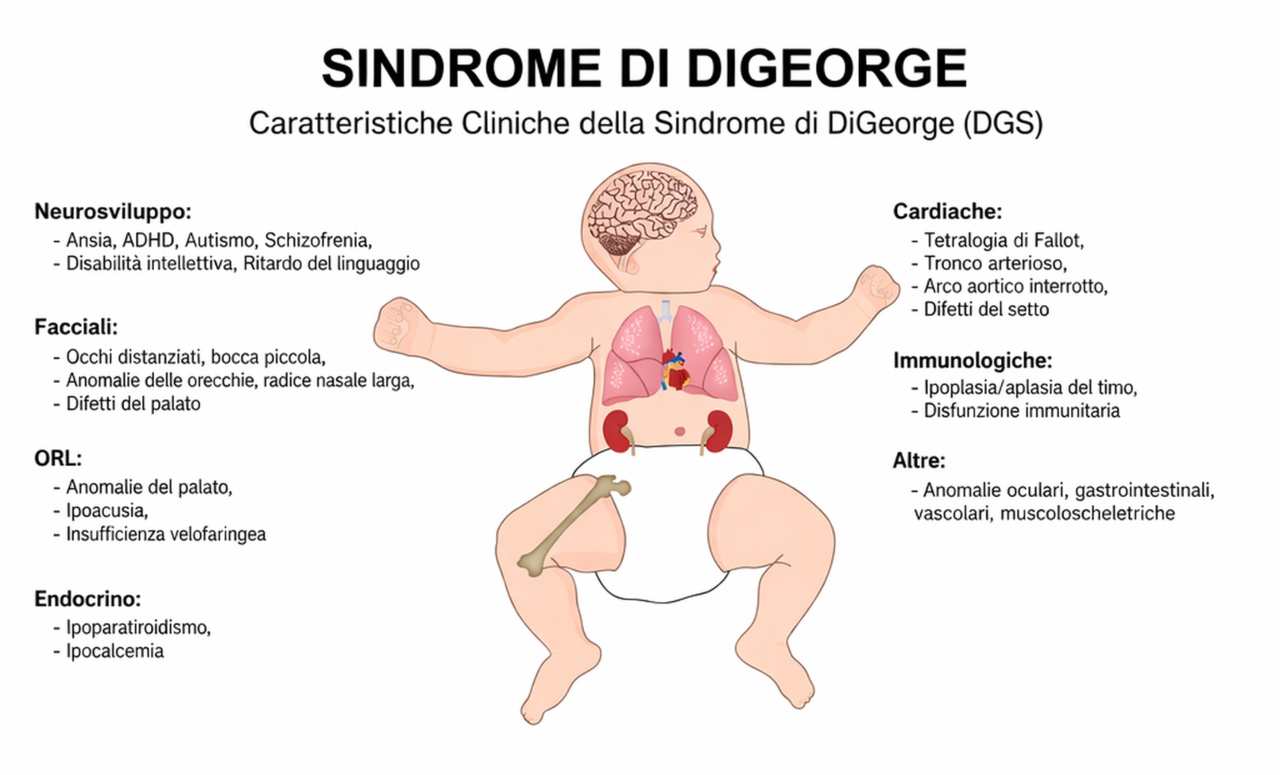
Il quadro clinico della sindrome di DiGeorge è uno dei più eterogenei in tutta la genetica clinica. Nessun singolo sintomo è presente in tutti i pazienti, e la gravità varia da forme praticamente asintomatiche a presentazioni che richiedono interventi chirurgici multipli nei primi mesi di vita.
Cardiopatie congenite
Le cardiopatie congenite sono la manifestazione più frequente e clinicamente rilevante nella fase neonatale. Si riscontrano in circa due terzi dei pazienti e comprendono prevalentemente malformazioni conotruncali, cioè difetti del tratto di efflusso del cuore e dell’arco aortico. Le più comuni sono: la tetralogia di Fallot (la forma più rappresentata, presente in circa il 36% dei soggetti con cardiopatia), il tronco arterioso, l’interruzione dell’arco aortico e i difetti del setto interventricolare. L’arco aortico destro, pur non sempre causa di compromissione emodinamica, si rileva in circa il 20% dei soggetti. La prognosi complessiva dipende in larga misura dalla gravità della cardiopatia e dalla tempestività dell’intervento chirurgico.
Anomalie del timo e deficit immunitario
Il timo è la ghiandola dove maturano i linfociti T, cellule fondamentali per l’immunità cellulare. Nella sindrome da delezione 22q11.2, il timo può essere ipoplasico (ridotto di dimensioni) o, in una minoranza di casi, completamente assente. Questo si traduce in un deficit della funzione linfocitaria T che varia enormemente da paziente a paziente.
Secondo la Prof.ssa Caterina Cancrini dell’Ospedale Bambino Gesù, un difetto lieve o moderato della funzione delle cellule T si riscontra nel 40-60% dei casi, mentre un quadro grave di immunodeficienza interessa solo lo 0,5-1% dei pazienti. La forma parziale espone i pazienti a infezioni ricorrenti, specialmente delle vie respiratorie, e aumenta il rischio di malattie autoimmuni: artrite idiopatica giovanile, anemia emolitica, pancitopenia autoimmune, tireopatie e vitiligine. La forma completa, in assenza di trattamento, è generalmente incompatibile con la sopravvivenza: richiede il trapianto di tessuto timico o di cellule staminali ematopoietiche, con risultati relativamente buoni in termini di ricostituzione delle cellule T nell’arco di 5-6 mesi (MSD Manuals professionale, citando Davies et al., J Allergy Clin Immunol 2017).
Ipoparatiroidismo e ipocalcemia
Le ghiandole paratiroidi regolano i livelli di calcio nel sangue attraverso la secrezione di paratormone. Nella Sindrome di DiGeorge, queste ghiandole sono spesso ipoplasiche o assenti, con conseguente ipoparatiroidismo e riduzione della calcemia. L’ipocalcemia si manifesta tipicamente nelle prime 24-48 ore dalla nascita con spasmi muscolari (tetania ipocalcemica) o convulsioni neonatali, e rappresenta spesso il primo segno clinico che porta al sospetto diagnostico nei bambini senza cardiopatia evidente. Nei casi parziali, l’ipoparatiroidismo è gestito con supplementazione di calcio e vitamina D.
Anomalie del palato e disturbi del linguaggio
Le anomalie palatali sono presenti in oltre il 65% dei pazienti e comprendono un ampio spettro: dall’insufficienza velofaringea (incapacità del velo palatino di chiudere adeguatamente il rinofaringe durante la fonazione), alla palatoschisi sottomucosa, all’ugola bifida, fino alla palatoschisi vera e propria nelle forme più gravi. La conseguenza più comune è una voce ipernasale e difficoltà di alimentazione nel neonato, legate a un’alterata coordinazione della suzione e della deglutizione, con possibile necessità di sondino nasogastrico nelle fasi iniziali. I disturbi del linguaggio rappresentano una delle manifestazioni più frequenti e diventano evidenti soprattutto dopo i due anni di età; possono associarsi, in alcuni casi, a ritardo dello sviluppo cognitivo o psicomotorio
Caratteristiche facciali
Le caratteristiche facciali della Sindrome di DiGeorge sono riconoscibili ma non sempre marcate. Il fenotipo tipico comprende un viso lungo e stretto, naso prominente con radice larga e punta bulbosa, mandibola ipoplasica, labbro superiore sottile e orecchie piccole, spesso dismorfiche, con padiglioni ripiegati e impianto basso. Possono essere presenti anche altre caratteristiche, come rime palpebrali strette, ptosi palpebrale o aumento della distanza interorbitale, sebbene in modo variabile. In molti pazienti questi tratti sono sfumati e possono passare inosservati e ciò contribuisce al ritardo diagnostico nei soggetti con presentazioni atipiche.
Disturbi del neurosviluppo e psichiatrici
Il coinvolgimento neurologico e psichiatrico è una componente sempre più riconosciuta della Sindrome di DiGeorge. La maggior parte dei pazienti presenta un ritardo variabile dello sviluppo, con difficoltà prevalenti nel linguaggio e nell’apprendimento. Sono comuni l’ADHD (disturbo da deficit di attenzione e iperattività) e i disturbi dello spettro autistico.
Come riportato da Sushma et al. 2025, nell’adolescenza e nell’età adulta, la sindrome si associa a un rischio aumentato di circa 20-30 volte di sviluppare schizofrenia rispetto alla popolazione generale, rendendola una delle varianti genetiche a più alto rischio per questa patologia. Il rischio di altre patologie psichiatriche, tra cui disturbo bipolare e disturbi d’ansia, è anch’esso aumentato. La riduzione dell’espressione del gene COMT, che degrada la dopamina nella corteccia prefrontale, è considerata uno dei meccanismi che contribuiscono a questa vulnerabilità neuropsichiatrica.
Altre manifestazioni sistemiche
Il quadro clinico si completa con una serie di manifestazioni che interessano altri apparati. Le anomalie renali (rene a ferro di cavallo, agenesia renale, rene multicistico) si riscontrano tra il 14 e il 35% dei pazienti. Il deficit uditivo, sia trasmissivo che neurosensoriale, è presente in proporzione simile e richiede una valutazione audiometrica precoce. Sono frequenti difficoltà di alimentazione nei primi mesi di vita (circa 30% dei casi), problemi di motilità gastrointestinale con tendenza alla stipsi, ipotiroidismo e trombocitopenia (riduzione delle piastrine). Un sottoinsieme di pazienti presenta anomalie scheletriche, oculari e delle vie aeree superiori.
Come si fa la diagnosi della Sindrome di DiGeorge

Il percorso diagnostico della Sindrome di DiGeorge parte quasi sempre da un sospetto clinico, che può nascere in epoche molto diverse: nel neonato con cardiopatia conotruncale o con crisi ipocalcemica nelle prime 48 ore di vita, nel bambino con infezioni ricorrenti e ritardo del linguaggio, nell’adolescente con voce ipernasale e difficoltà di apprendimento mai spiegate, o nell’adulto con psicosi a esordio precoce.
La conferma diagnostica si ottiene con tecniche citogenetiche o molecolari in grado di identificare la delezione 22q11.2. Le metodiche attualmente raccomandate includono (ISSalute; Orphanet):
- MLPA (Multiplex Ligation-dependent Probe Amplification): tecnica quantitativa con buona copertura della regione critica.
- Array-CGH (Comparative Genomic Hybridization su array) e SNP microarray genome-wide: metodiche ad alta risoluzione che consentono di identificare delezioni di dimensioni variabili su tutto il genoma, incluse delezioni atipiche o annidate nella regione 22q11.2.
- FISH (Fluorescence In Situ Hybridization): tecnica storica, specifica per la regione 22q11.2 ma limitata nella risoluzione e oggi meno utilizzata come test di primo livello.
La diagnosi molecolare è fondamentale non solo per confermare il caso indice, ma anche per studiare i genitori poiché in caso di delezione de novo la probabilità di ricorrenza nei fratelli è bassa (circa 2-3%, legata a un possibile mosaicismo germinale di basso grado), mentre in caso di delezione ereditata da un genitore portatore la probabilità sale al 50% per ciascuna gravidanza successiva.
In Italia, la sindrome da delezione 22q11.2 è inclusa nell’elenco delle malattie rare esenti dal ticket (DPCM 12 gennaio 2017, allegato 7) con il codice RCG160, afferente al gruppo delle immunodeficienze primarie. I centri specializzati per la diagnosi e la presa in carico sono accessibili attraverso il Telefono Verde Malattie Rare (800.89.69.49) dell’Istituto Superiore di Sanità, attivo dal lunedì al venerdì dalle 9 alle 13, come riportato da ISSalute. A livello europeo, la sindrome rientra nella rete ERN-Rita (European Reference Network per i disturbi immunologici rari).
Diagnosi prenatale con metodi invasivi
Quando una coppia ha già avuto un figlio affetto, o quando uno dei genitori è risultato portatore della delezione 22q11.2 a seguito di uno studio familiare, la diagnosi prenatale è indicata e possibile con metodiche invasive. Le opzioni disponibili sono la villocentesi (prelievo dei villi coriali, eseguibile dalla 11a settimana) e l’amniocentesi (dalla 15a-16a settimana), entrambe in grado di fornire materiale fetale per l’analisi molecolare della delezione mediante tecniche ad alta risoluzione, come array-CGH o SNP microarray.
In caso di cardiopatia conotruncale fetale evidenziata all’ecocardiografia di secondo livello, anche in assenza di familiarità nota per la sindrome, la diagnosi prenatale invasiva può essere proposta perché le cardiopatie conotruncali sono frequentemente associate alla Sindrome di DiGeorge. Per le coppie che ricorrono alla procreazione medicalmente assistita, è disponibile anche la diagnosi genetica preimpianto (PGT-M), che consente di selezionare embrioni privi della delezione prima del trasferimento in utero.
Gestione clinica e trattamento della Sindrome di DiGeorge
Non esiste una cura per la Sindrome di DiGeorge. La gestione si basa su un approccio multidisciplinare e sintomatico, orientato a trattare le specifiche manifestazioni presenti in ciascun paziente e a prevenire o monitorare le complicanze più comuni.
Le cardiopatie congenite richiedono spesso correzione chirurgica nelle prime settimane o mesi di vita; i tempi e le modalità dipendono dal tipo di difetto e dalla sua gravità. L’ipocalcemia da ipoparatiroidismo viene trattata con supplementazione di calcio e vitamina D nelle forme parziali. Il deficit immunitario grave nelle forme complete richiede il trapianto di tessuto timico o, in alternativa, di cellule staminali ematopoietiche. Le anomalie palatali con insufficienza velofaringea possono richiedere intervento chirurgico o terapia protesica, sempre accompagnata da logopedia intensiva.
Il follow-up a lungo termine prevede monitoraggi periodici della funzione immunitaria, della calcemia e della funzione tiroidea, valutazioni audiometriche cadenzate, supporto logopedico e psicopedagogico, valutazione neuropsichiatrica nell’adolescenza e monitoraggio cardiologico per tutta la vita. L’aspettativa di vita è variabile e dipende in larga misura dalla gravità delle manifestazioni, in particolare cardiache e immunologiche; tuttavia, molti pazienti che raggiungono l’età adulta possono condurre una vita relativamente autonoma, pur con necessità di supporto in alcuni casi.
Prenatalsafe 5DiGeorge: lo screening prenatale non invasivo per la Sindrome di DiGeorge

Nel contesto clinico appena descritto, la diagnosi prenatale della sindrome da delezione 22q11.2 ha assunto un ruolo crescente negli ultimi anni, reso possibile dallo sviluppo dei test di screening prenatale non invasivo (NIPT) basati sull’analisi del DNA fetale libero circolante nel sangue materno. Tra questi, il Prenatalsafe 5DiGeorge, sviluppato da Eurofins Genoma, è la versione del test Prenatalsafe specificamente progettata per includere lo screening della microdelezione del cromosoma 22 (22q11.2).
Il test analizza il DNA fetale libero circolante (cfDNA) isolato dalla componente plasmatica del sangue materno, attraverso il sequenziamento massivo parallelo (MPS) su piattaforme NGS Illumina e Thermofisher, integrato dall’algoritmo proprietario NIPT Flow™, con tecnologia certificata CE IVD. Oltre alla ricerca della microdelezione 22q11.2 (sindrome di DiGeorge), il Prenatalsafe 5DiGeorge valuta le aneuploidie dei cromosomi 21 (sindrome di Down), 18 (sindrome di Edwards), 13 (sindrome di Patau) e le aneuploidie dei cromosomi sessuali, con possibilità di determinazione del sesso fetale su richiesta.
Il test può essere eseguito a partire dalla 10a settimana di gravidanza con un semplice prelievo di sangue venoso materno. Il referto è disponibile in circa 7 giorni dalla ricezione del campione al laboratorio.
Quando è indicato il Prenatalsafe 5DiGeorge
Il test può essere considerato in alcune situazioni cliniche, come la presenza di anomalie conotruncali all’ecografia fetale, un rischio aumentato agli screening del primo o secondo trimestre, o quando la coppia non desidera ricorrere a metodiche invasive. In caso di familiarità nota per delezione 22q11.2, tuttavia, la diagnosi prenatale invasiva rimane la scelta raccomandata.
È importante precisare che il Prenatalsafe 5DiGeorge, come tutti i NIPT, è un test di screening e non di diagnosi. Infatti, un risultato ad alto rischio richiede conferma con una metodica diagnostica invasiva (villocentesi o amniocentesi). Un risultato a basso rischio riduce significativamente la probabilità della condizione, ma non la esclude con certezza assoluta. Il test non è prescrivibile per le gravidanze gemellari bicoriali.
Consulenza genetica integrata
Il percorso Prenatalsafe include una consulenza genetica pre-test e post-test gratuita, fondamentale per contestualizzare il risultato, spiegare i limiti e le potenzialità del test e pianificare eventuali approfondimenti. In caso di risultato positivo, il programma prevede anche la possibilità di un follow-up gratuito con villocentesi o amniocentesi presso ginecologi di riferimento convenzionati con Eurofins Genoma, garantendo alla coppia un percorso diagnostico completo e supportato.
Per chi si trova di fronte alla complessità della sindrome da delezione 22q11.2 e vuole capire quali opzioni di screening prenatale sono disponibili nella propria gravidanza, il punto di partenza è sempre una consulenza con il proprio ginecologo o con un genetista clinico, che potrà valutare il profilo di rischio individuale e indicare la versione di Prenatalsafe più adatta al caso specifico.
